research interests
overview
The primary goal of the Levine Research Group at the Yale School of Medicine (LRG@YSM) is to understand and intervene in protein folding diseases using biophysical insights. In particular, we strive to understand the underlying physical and thermodynamic mechanisms that lead to age-related disease in a diverse set of biological environments. We focus predominantly on intrinsically disordered proteins (IDPs) and regions (IDRs), which tend to interconvert between three-dimensional structures. These behaviors enable IDPs to carry out diverse physiological roles, however it can also lead to the aggregation of soluble and insoluble protein plaques (amyloids) over the lifecourse, which is a notable hallmark in certain cancers and in over twenty-one degenerative diseases such as Alzheimer's Disease, Parkinson's Disease, and Type II Diabetes. To better understand these behaviors, LRG@YSM utilizes a combination of molecular dynamics simulations and experiments that combine biophysical, biochemical, biological, and clinical knowledge in order to manipulate proteotoxic species in cancer, human aging, and in traditional amyloid diseases.


molecular modeling
High-performance computing and molecular dynamics (MD) simulations enables us to recreate chemical and biological systems from first-principles, providing nanoscale determinants of proteostasis. Parallelization of simulations also allows for the folding of unstructured proteins, which are notoriously difficult to characterize from experiments alone. Our lab employs many theoretical techniques, including replica-exchange MD (REMD), umbrella sampling, thermodynamic integration, and free energy perturbations.
Solution Biophysics Experiments
Targeting Soluble amyloids in Age-Related Disorders
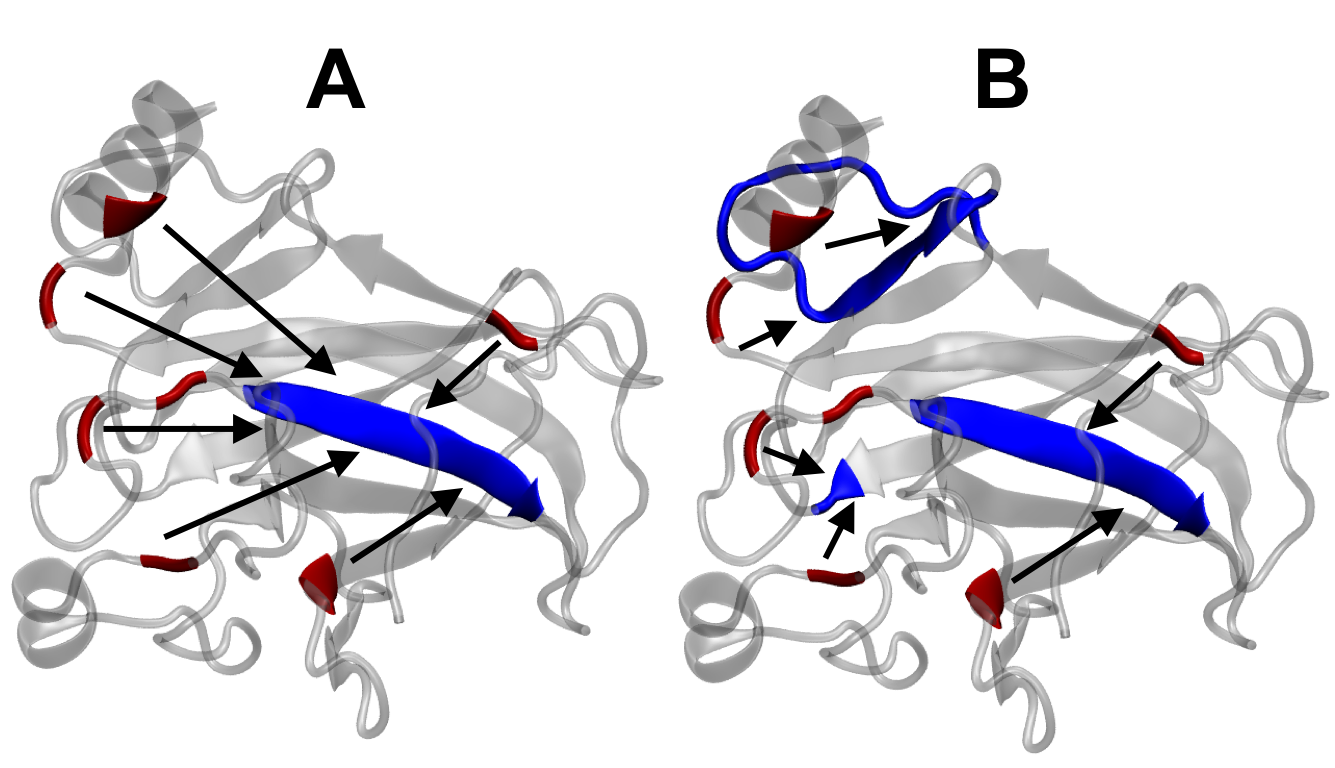
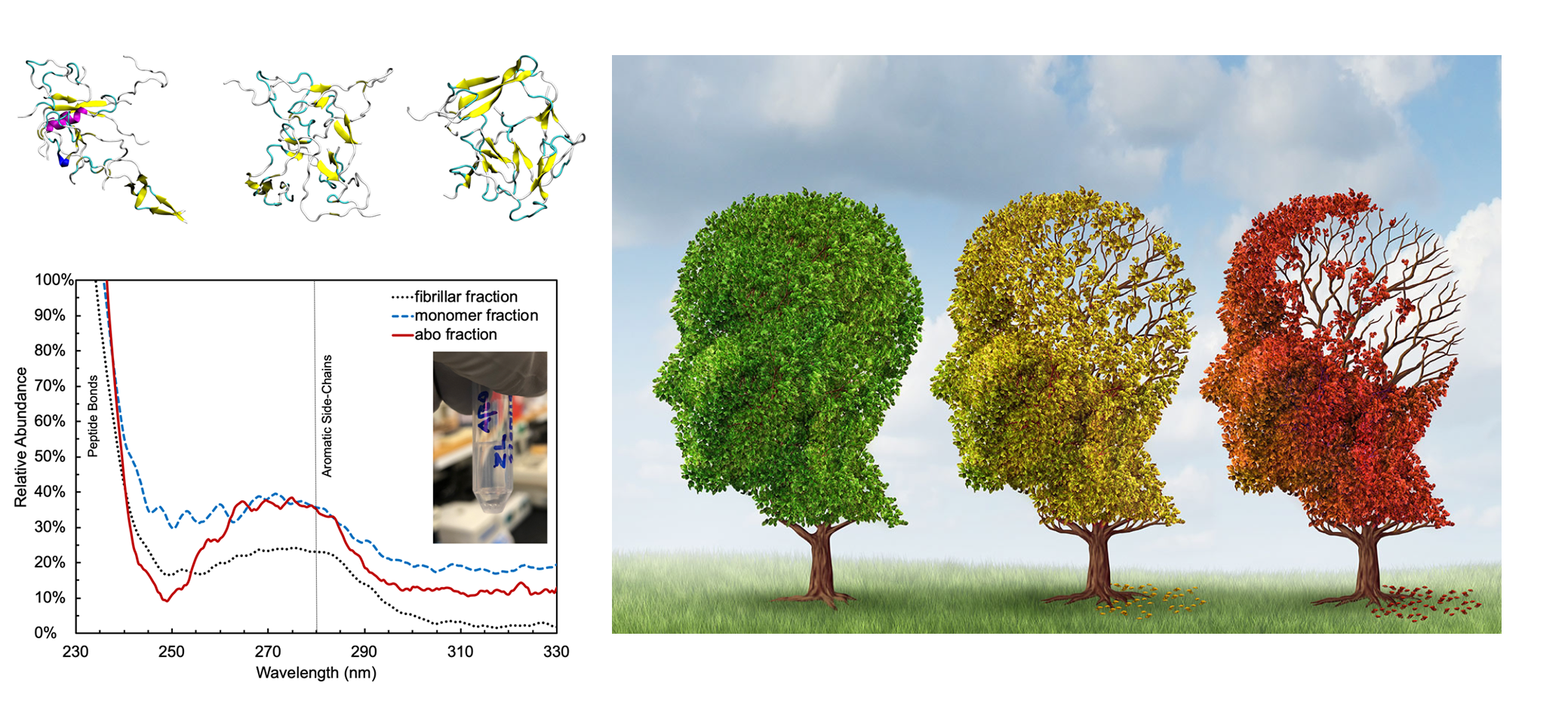
Cognitive decline in Alzheimer’s Disease (AD) and many other age-related dementias have long been associated with the presence of insoluble amyloid plaques that disrupt normal synaptic functioning. However, more recent studies have revealed that cellular impairment is much more potently associated with soluble protein oligomers rather than from insoluble fibrils. Soluble oligomers are structurally irregular and promiscuously bind to membrane proteins, thereby dysregulating downstream processes. This heterogeneity, however, makes it difficult to experimentally characterize transient oligomers, despite their tremendous importance.
To address these challenges, we combine molecular simulations and single-molecule experiments to probe the distribution of individual oligomers and their structural and functional properties. We also target protein oligomers using ssDNA and peptide aptamers to track and neutralize promiscuous protein species in cell lysates. This work bridges basic biophysical insights with macroscopic cellular behaviors to inform how soluble amyloid species drive cellular aging and multiple degenerative disorders.
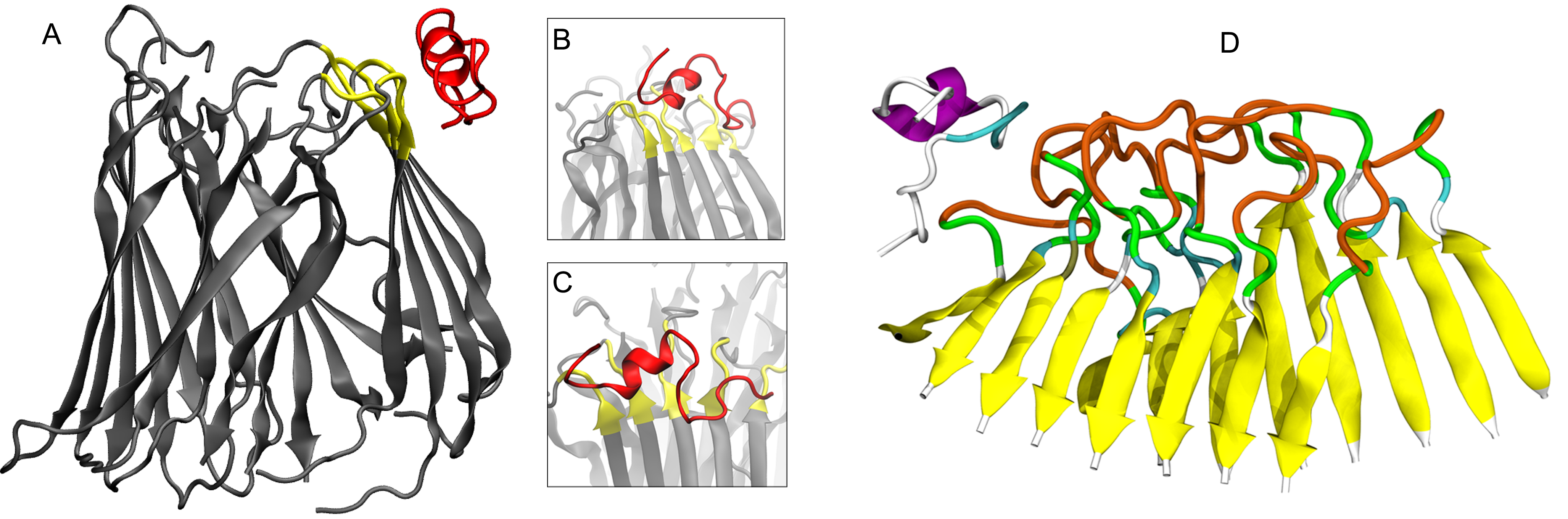
Regulation of Tumor Suppression Proteins using Amyloid-Disrupting Peptides
In over half of all cancer types, tumor suppressor proteins such as p53 can become inactivated by single amino acid mutations, inhibiting their ability to induce programmed cell death. Follow-up studies have indicated that misfolded tumor suppressor proteins can outwardly display sticky amino acids that increase their aggregation propensity to one another and subsequently disrupt their ability to function. Thus, many cancers can be thought of as protein aggregation diseases where protein regulators are no longer able to suppress aberrant behaviors.
Furthermore, many of the most lethal cancer types such as high-grade serous ovarian carcinomas (HGSOCs) exhibit mutations that induce aggregation in over 96% of cases. While there are currently no therapies to address protein co-aggregation in cancer, there is emerging evidence that amyloid-inhibiting peptides may potently inhibit p53 aggregation in HGSOCs and related tumors. This suggests that targeting the protein aggresome can significantly enhance tumor treatment plans and rescue paralog tumor suppressor proteins from co-aggregation, thereby laying the foundation for next-generation cancer therapeutics.